
ENSURING STUDY APPROVAL UNDER EU MDR REGULATION FOR MEDICAL DEVICE CLINICAL TRIALS
This is a comprehensive review of the process, pitfalls, and procedures to get study approval for medical device clinical trials. We look into the future and unpack the present, in terms of European medical device regulation and the local regulatory landscape. Along with the intricacies of EU MDR 2017/745 medical device regulations, we provide an important summary of medical device study classifications and the corresponding submission pathways.
Classification and initial submission for medical device clinical trials under EU MDR regulation – EU MDR 2017/745
Under the European Medical Devices Regulation (EU) 2017/745 (MDR), there are different frameworks for medical device clinical trials. European medical device regulation allows each to have its own regulatory pathway, as shown in Figure 1 below:
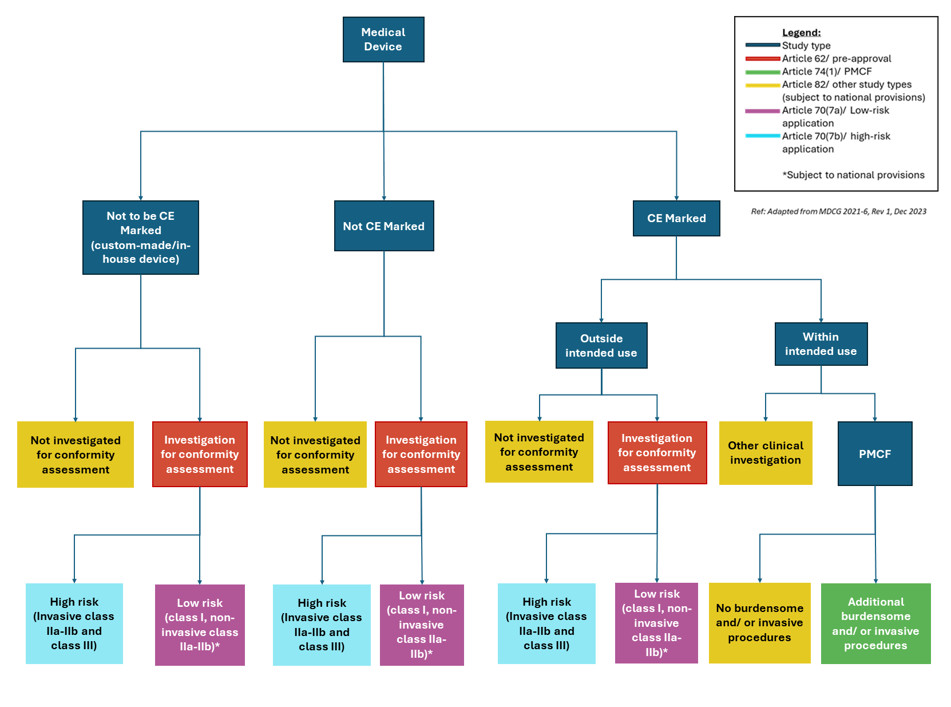
Validation and Assessment – Pre-approval studies (Art. 62)
Medical device clinical trials demonstrating safety and/or performance to support their conformity assessment, fall within Article 62 of the EU MDR regulation. Their regulatory pathways can differ depending on the medical device classification.
Low-Risk Devices
Medical device clinical trials for low-risk devices (class I and non-invasive class IIa and IIb) can start after validation. This is under Article 70 (7)(a) and is subject to national provisions and a positive ethics opinion. The validation process can take at least 10 days but up to 25 days with queries. In rare circumstances, the process can take up to 55 days if all the relevant extensions are applied.
High-Risk Devices
Medical device clinical trials for high-risk devices (invasive class IIa, invasive class IIb, and class III) can only start after a full assessment of the application and receipt of a positive ethics opinion. After the validation process described above, there is an additional assessment period. This can take between 45-65 days, excluding any postponement caused by a ‘clock stop’ during a query response.
Post-Market Clinical Follow-Up (PMCF)
Under Article 74 (1), post-market clinical follow-up (PMCF) studies which include procedures additional and burdensome to those performed under normal conditions of use of the device are subject to a 30-day notification procedure. The requirements of PMCFs are similar to pre-approval studies (i.e. documentation, substantial modifications, legal representation and informed consent).
Sponsors of clinical investigations (including PMCFs) outside the EU must have a legal representative in the EU who ensures the sponsor complies with EU MDR regulations. In addition, manufacturers should ensure they have a Person Responsible for Regulatory Compliance (PRRC) for medical device clinical trials. (Article 15 (3)(e)).
Lumis Tip
Classify your study early to understand regulatory requirements. Document your classification.
EUDAMED
A EUropean DAtabase on MEdical Devices (EUDAMED) is in the process of development in accordance with Article 73. This will be an online database where sponsors will submit applications for clinical investigations and report clinical investigation safety events. EUDAMED will also be used in other product lifecycle stages.
Once functional, EUDAMED will have six modules, one of which will be for clinical investigations and performance studies (CI/PI). However, this CI/PS module is not scheduled for completion until Q3 2026 and is then subject to an audit – after which it will become mandatory in Q4 2027. Until then, clinical investigation applications must be made at a national level. Individual countries may have different submission procedures, therefore, thorough research and planning at a national level is required – particularly for multi-national studies.
Lumis Tip
Without EUDAMED, submissions are at a national level. Use national CA and EC websites to understand regulatory requirements for each country.
Documentation Requirements
Pre-approval clinical investigations (Article 62) and post-market clinical follow-ups (Article 74) are subject to the same documentation requirements as outlined in Chapter II, Annex XV of the EU MDR.
These requirements are:
- Application form
- Investigator Brochure (IB)
- Clinical Investigation Plan (CIP)
- Other information
- GSPR Statement
- Ethics committees’ (EC) opinion, when applicable
- Insurance
- Participant documents (i.e. informed consent and information sheet)
- Data protection arrangements (if not in any of the above documents – you could include it in a cover letter)
MDCG 2021-08 contains templates for the application form, GSPR checklist, and other documentation. ISO 14155:2020 and the EU MDR provide further details of what is expected in certain documents, e.g. the CIP and IB. However, because national competent authorities (CA) may request further documentation (e.g. risk management) and have local document requirements (e.g. insurance) it is recommended to consult their websites before submission.
Medical device clinical trials not performed to support the device’s conformity assessment or as a PMCF, as mentioned in Article 82(2), are subject to national procedures.
For example, in Austria the competent authority, BASG, states that the procedure followed depends on the clinical consequence. Article 82 studies that have therapeutic or diagnostic consequences are subject to a full authorisation (i.e. Article 70 (7) (b)). Studies without any clinical consequence should be notified to BASG 30 days in advance, like Article 74(1).
In Ireland, the HPRA just review Article 82 studies like a PMCF – i.e. a notification is required 30 days before the study starts. The documentation requirements are also subject to national requirements.
Maintaining compliance with EU MDR 2017/745
Substantial modifications are defined as any change to medical device clinical trials or a PMCF medical device that significantly impacts the safety or rights of a participant or the validity or quality of data collected.
For example:
- Changes to study endpoints,
- Change in treatment regime,
- Change in risk/benefit ratio from new preclinical data,
- Change in IFU, etc.
You can refer to MDCG 2021-6 Annex II for a list of examples.
Lumis Tip
Document the rationale why a given amendment is substantial/non-substantial.
A substantial modification submission would consist of:
- A cover letter,
- An application form,
- Amended documents,
- Possibly, evidence of fee payment.
You must carefully consider which documents to update. For example, if you update the CIP this may impact the Participant Information Sheet and Informed Consent documents. Therefore, you may have to submit the modification to the competent authority and/or ethics committee.
Lumis Tip
Due to the burden of national amendment submissions, try and group amendments together to reduce regulatory workload.
Until EUDAMED is active, substantial modifications should be submitted at a national level. For example, in Germany, it’s via the DMIDS database. The CA has 37 days to review the application and issue a rejection or request for information. An additional 7 days can be added by the CA if experts must be consulted.
Ethics committees generally follow these rules but as they are managed at a national level, it is best to check their websites for further details. Non-substantial amendments are managed under national regulations so check the relevant CA and EC websites.
The EU MDR regulations and guidance do not mention Urgent Safety Measures (USMs), however, there may be national provisions for these situations. For example, Ireland NREC-MD permits USMs but they should be reported within 3 days. Austria also has its own form that must be used to notify the CA – notification to the EC would also be required separately. Of course, there are also safety reporting requirements (Article 80) for clinical investigations but the reporting requirements for PMCFs are different.
Annual reporting is not a requirement under the European medical device regulation but still may be required by some member states’ ethics committees and/or CA.
Lumis Tip
There can be national requirements additional to those described in the MDR (e.g. annual reporting and USMs).
End of trial & EU MDR regulation
The sponsor must notify a member state (MS) within 15 days of the end of the medical device clinical trial or PMCF in that country (“local end of trial”). For studies across multiple MSs, the sponsor must notify all MSs within 15 days of the trial’s completion in the final MS (“EU end of trial”).
For studies that continue in third countries after the EU end-of-trial, sponsors should inform the MSs when the “global end-of-trial” is forecasted, as this impacts the clinical investigation report deadline. For example, this could be done via a cover letter to MSs during “EU end of trial”. Sponsors are advised to notify MSs once the study ends globally.
If a sponsor decides their clinical investigation should be temporarily halted or terminated early, they have 15 days to inform concerned MSs (or 24 hours if based on safety grounds). A notification will be needed to restart the study – a substantial modification is needed if the temporary halt is for safety reasons.
Lumis Tip
There can be multiple end-of-trial submissions (e.g. “local”, “EU”, and “global”).
Reporting within the framework of EU MDR regulation
A clinical investigation report (CIR) should be submitted within one year of the global end of the clinical investigation/ PMCF to all concerned MSs. Studies subject to early termination or temporary halts have 3 months to submit the CIR – a risk analysis is required for temporary halts due to safety.
If the clinical investigation restarts within 3 months, a CIR is not needed until the end of the study but this CIR should include details on the temporary halt. The contents of the CIR are outlined in the MDR (Section 7, Chapter III of Annex XV) and ISO 14155:2020 (Annex D).
Furthermore, a CIR summary, written to be understood by the intended user, who could be a layperson, should accompany the CIR (see the Commission’s guidance 2023/C 163/06 for further information on the summary CIR’s content). Note that both the CIR and its summary will be made publicly available once EUDAMED is active.
Submission procedures for end-of-trial and CIR submission are at a local level and sponsors should consider procedures for both the CA and EC.
Special Circumstances for Medical Device Clinical Trials
Combination studies
The regulatory procedures vary depending on the drug-device design and member state requirements. European guidance makes the distinction between combination products where:
- the medicine and device are integrated and cannot be separated (e.g. transdermal patch) -referred to as “integral” products.
- the medicine and device are physically separate (e.g. nebuliser vs a bottle of medicine it vaporises) referred to as “non-integral” products2,6.
For studies investigating non-integral combination products, the study falls under both the MDR (for the device component) and Clinical Trials Regulations (CTR) (for the medicine’s component).
Integral products’ primary mode of action determines the regulatory framework. If the principal mode of action is the medicinal product, the study falls within the CTR. If, however, the principal mode of action is the device, the study falls within the MDR.
If the product is an integral combination where the medicinal product is the principal mode of action an MDR submission isn’t required. However, compliance with General Safety and Performance Requirements is still obligated and sometimes further documentation is needed for submission (e.g. Ireland’s NREC).
For other combination products, typically two submissions are required: one under CTR and the second under MDR. It is recommended to refer to this joint application in the cover letter for both the MDR and CTR submissions. Ideally, both MDR and CTR submissions are made simultaneously so any updates in documents can be done for both applications at the same time.
If a study falls under the MDR and CTR, the sponsor must be familiar with the requirements of both legislations (e.g. amendments and safety reporting). Due to the administrative burden of combination studies, plan amendment submissions strategically.
Consult with the relevant CAs & ECs to understand combination study requirements, as they can vary by member state.
Lumis Tip
When submitting under the MDR and CTR, refer to the joint application in the cover letters for both submissions.
Radiation
Some European countries require additional documentation or procedures if a clinical study will use radioactive substances or ionizing radiation (e.g. Germany, UK, and Ireland). These procedures normally require specialists (medical physicists and/or clinical radiologists) to provide input to the documentation. If your study involves ionizing radiation or radioactive substances, check the member state requirements early and, if necessary, seek the relevant experts.
Other
Documentation and procedural requirements can be impacted by study populations (e.g. pediatrics, emergency situations), product type (e.g. ATMP-Device combination), and study design (e.g. decentralized trial).
In conclusion
The European Medical Devices Regulation has changed how medical device study approvals are obtained and understanding the study’s classification and submission planning is required early on. During this planning, sponsors must consider any national requirements (e.g. documentation or maintenance submissions, like reporting) as well as any special circumstances, like studying combination products or using radiation in a study. Only after applying this holistic approach can a sponsor ensure compliance.
If you need any support with getting your medical device study approval in Europe, contact us today!
References
- Medical Devices Regulation (EU) 2017/745, 5 April 2017, Amended 20th March 2023. https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:02017R0745-20230320
- MDCG 2021-6 Rev. 1 Regulation (EU) 2017/745 – Questions & Answers regarding clinical investigation December 2023. https://health.ec.europa.eu/system/files/2023-12/mdcg_2021-6_en.pdf
- MDCG 2021-08. Clinical investigation application/notification documents. May 2021. https://health.ec.europa.eu/system/files/2021-05/mdcg_2021-8_en_0.pdf
- ISO 14155:2020 Clinical investigation of medical devices for human subjects Good clinical practice. https://www.iso.org/standard/71690.html
- Commission Guidance on the content and structure of the summary of the clinical investigation report (2023/C 163/06). https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:52023XC0508(01)
- Eudralex Volume 10 – Guidance documents applying to clinical trials, Questions & Answers, Version 6.7. December 2023. https://health.ec.europa.eu/system/files/2023-12/regulation5362014_qa_en_0.pdf
