
Transitioning studies approved under the Clinical Trials Directive 2001/20/EC (CTD) to the new Clinical Trials Regulation 536/2014 (CTR) is a common challenge for sponsors. This blog will provide an overview of how to transition a study to the CTR and some key considerations.

What clinical trials must transition to the Clinical Trials Regulation?
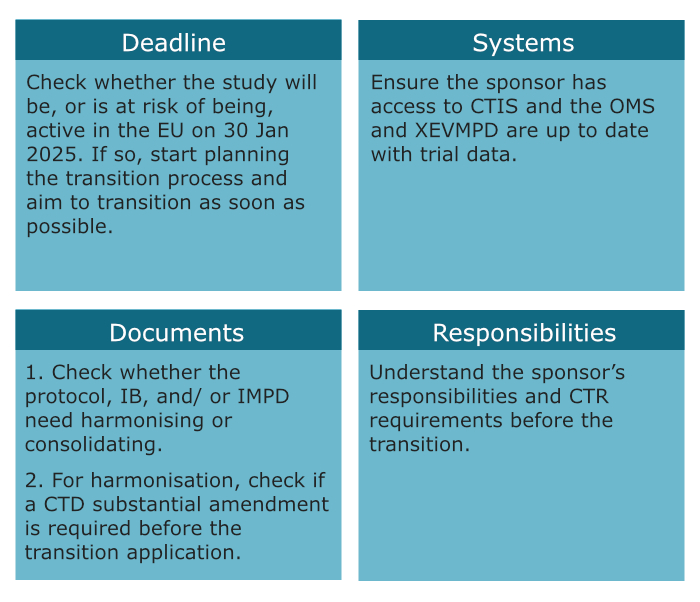
Clinical trials approved under the Clinical Trials Directive with at least one site in the European Union forecasted to remain active on 30 Jan 2025 must be transitioned1. This means sponsors must carefully assess whether their study will remain active on 30 Jan 2025. Even if a study is scheduled to finish before the 30 Jan 2025, the sponsor must consider any risk of delays and whether this may warrant transitioning to the CTR – regular assessments of study timelines are recommended.
How is a transition application submitted?
A transition application is submitted via the Clinical Trials Information System (CTIS). This system interacts with the eXtended EudraVigilance Medicinal Product Dictionary (XEVMPD) and Organisation Management System (OMS). Therefore, it is important the investigational product(s) are registered in XEVMPD and the applicable organisations (e.g. sponsor) are registered in OMS.
The documents needed for submission should be already approved under CTD and will consist of part I documents (e.g. protocol, investigator brochure, GMP documents, etc.) and part II documents (i.e. participant information sheet and informed consent form). This is additional to a cover letter, GDPR statement, and a CTIS application form1.
For multinational trials under the CTD, the protocol, investigator brochure (IB), and investigational medicinal product dossier (IMPD) can vary between member states. Consequently, sponsors must “harmonise” or “consolidate” said documents – this involves updating the study documents which may require a CTD substantial amendment2,3. These document updates may impact other study documents, like the participant information sheet and informed consent (PIS-ICF). Sponsors must also prepare redacted documents for the transition1.


When the trial is entered into CTIS and documents uploaded, the application can be made and the review period is up to 60 days with a maximum of 106 days (potentially more for some investigational product types)4,5. It is possible to submit a minimal document set which can result in an expedited approval of 22 days; however, this will impact what must be submitted under the first substantial modification1.
When should a trial transition?
The deadline to transition clinical trials to the Clinical Trials Regulation is 30 Jan 2025, however, we recommend sponsors aim to transition their studies as soon as possible. Some studies will require study document updates and a CTD substantial amendment before the transition application. Additionally, time should be factored in to prepare the transition application and up to 106 days for the study to be approved under Clinical Trial Regulation (sometimes longer).
What happens after the transition?
Trials will be regulated under the Clinical Trials Regulation 536/2014 when the transition application is approved. Therefore, sponsors must comply with Clinical Trials Regulation requirements (e.g. safety reporting, archiving, transparency) even if these are not mentioned in the protocol. At the first Clinical Trials Regulation substantial modification, the sponsor must align the study documents (incl. labels) with the CTR and submit any remaining documents for part I and part II (including redacted documents).
What are the key considerations?

How can Lumis Consulting help?
Lumis Life Science Consulting supports pharmaceutical companies by providing solutions in regulatory affairs and clinical trial processes and oversight. We understand the complexities of the Clinical Trials Regulation 536/2014 and have a team of experienced professionals with deep industry expertise. Therefore, Lumis is well-positioned to provide strategic guidance and operational services tailored to the needs of the sponsor and study.
Contact UsReferences
- Guidance for the Transition of clinical trials from the Clinical Trials Directive to the Clinical Trials Regulation. 21 Dec 2023. https://health.ec.europa.eu/system/files/2023-12/transition_ct_dir-reg_guidance_en.pdf
- CTCG Best Practice Guide for sponsors of multinational clinical trials with different Part I document versions approved in different Member States under the Directive 2001/20/EC that will transition to the Regulation (EU) No. 536/2014 . Nov 2023. https://www.hma.eu/fileadmin/dateien/HMA_joint/00-_About_HMA/03-Working_Groups/CTCG/2023_11_CTCG_Best_Practice_Guide_for_sponsors.pdf
- CTFG Best Practice Guide for sponsors of multinational clinical trials with different protocol versions approved in different Member States under Directive 2001/20/EC that will transition to Regulation (EU) No. 536/2014. April 2018. https://www.hma.eu/fileadmin/dateien/Human_Medicines/01-About_HMA/Working_Groups/CTFG/2018_05_CTFG_Best_Practice_Guide_for_sponsors_of_transition_multinational_clinical_trials.pdf
- CTIS Training Programme – Module 23- Transitional trials from EudraCT to CTIS. Jan 2023. https://www.ema.europa.eu/en/documents/other/faqs-transition-trials-eudract-ctis-ctis-training-programme-module-23_en.pdf
- Clinical Trials Information System (CTIS) – Sponsor Handbook. 6 Nov 2023. https://www.ema.europa.eu/en/documents/other/clinical-trial-information-system-ctis-sponsor-handbook_.pdf
